| Key Learning Goals for this Lesson: |
|
For many of our analyses, we did a test for each feature. However, for clustering and classification, we used a subset of the features simultaneously. Methods that use multiple features are called multivariate methods and are the topic of this chapter.
Multivariate methods may be supervised or unsupervised. Unsupervised methods such as clustering are exploratory in nature. They help you find patterns that you didn't know were there and may also help you confirm patterns that were you knew were there. Supervised methods use some type of response variable to discover patterns associated with the response.
Unsupervised methods often work reasonably well even without replication in the data (as long as there are multiple conditions). Basically, they organize the data matrix into clusters of rows (features) or columns (samples) that have similar patterns. The features or samples in the clusters then serve as if they were replicates. We have already seen an example in cluster analysis, where we clustered the differentially expressing genes. We can see the dominant expression pattern in each cluster, and perhaps summarize it by averaging the component gene expressions for each treatment.
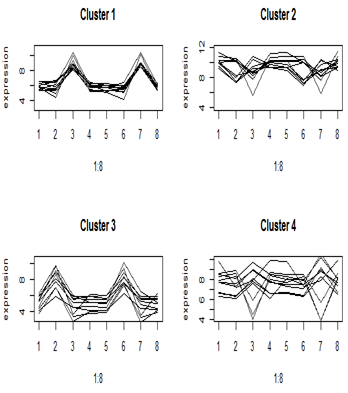
Here are the cluster profiles that we got from four of the eight clusters that we pulled out of the primate brain data using Euclidean distance. There were four brain regions and two species, giving eight treatment means. Clusters 1 and 3 show some clear patterns in gene expression, but clusters 2 and 4 appear to be genes with similar mean expression levels.
One-variable-at-a-time analyses such as t-tests and Fisher’s exact test do not take advantage of patterns across genes (although we have borrowed information from all of the genes to get estimates of the variance). For each gene we did a test of whether was differentially expressed. These are therefore called univariate methods.
Multivariate methods like clustering take advantage of the similarities among the features to reduce the data to a smaller number of patterns (sometimes called "eigenfeatures"). For some analyses, we prefer to take advantage of the similarities among the samples to obtain "eigensamples". Since there are fewer eigenfeatures than features, and fewer eigensamples than samples, we call these methods "dimension reduction" methods.
Dimension reduction is a set of multivariate techniques that find patterns in high dimensional data. Many commonly used dimension reduction methods are simple decompositions of the data matrix into a product of simpler matrices.
Dimension reduction methods come in unsupervised and supervised forms. Unsupervised methods include the singular value decomposition (SVD) and principal components analysis (PCA) which use only the matrix of features by samples as well as clustering. Supervised methods include multiple regression and classification, as well as more recently developed techniques such as sliced inverse regression (SIR) and require a response variable, which is usually a phenotype, in addition to the feature by sample matrix.
The most fundamental dimension reduction method is called the singular value decomposition or SVD. Oddly, statisticians don't seem to know much about this (although we use a related method, principal components analysis, very frequently). However in computer science and machine learning, SVD is one of the most important computational methods.
Our data consist of n samples and g feature values arranged as a g × n matrix which I will call M. (I will assume here that g>n but if not, the matrix can be transposed.) The features might be genes or protein binding sites, and the values are the normalized expression values. It is important to use the normalized values when using these dimension reduction methods. For microarray data, the values will be the usual normalized data. For sequencing data, the pseudo-counts computed by multiplying the actual counts by the normalization factors are used to normalize the samples to an equivalent library size. If the data are not normalized, the patterns that are extracted are often specific to the samples with the higher values, rather than being typical of the data as a whole.
Any g × n matrix M can be decomposed into 3 matrices U, D, V where
\[M = UDV^T\]
and:
The columns of U are \(U_1, U_2, \cdots U_n\). \(U_i\) is the ith eigensample. Each eigensample has an entry for each of the \(g\) features. Similarly the columns of \(V\) are \(V_1, V_2, \cdots V_n\). \(V_i\) is the ith eigenfeature. Each eigenfeature has an entry for each of the n samples. The normalized singular values \(d_{ii}^2/ \sum_{j=1}^n d_{jj}^2 \) have an interpretation as the relative size of the pattern described by eigenfeature and eigensample i and are sometimes called the percent variance explained by the eigencomponents.
Another way to look at the decomposition of M which helps us to understand why the eigencomponents pick out patterns is
\[M=\sum_{i=1}^n d_{ii} U_i V_i^\top \]
This says that M is the sum of matrices each of which is the product of one eigensample and one eigengene, and weighted by the corresponding singular value. Since all the eigensamples and eigengenes have norm one, the size of the component matrix is determined by the singular values. So the first few components "explain" most of the patterns in the data matrix.
To see how this works, we will look at the primate brain data which we used in homework 5.
Principal Components Analysis
The SVD is a matrix decomposition, but it is not tied to any particular statistical method. A closely related method, Principal Components Analysis or PCA, is one of the most important methods in multivariate statistics.
Suppose M is the data matrix. The column-centered data matrix \(\tilde{M}\) is computed by subtracting the column mean from the entries of each column. The principal components of M are the eigenvectors of \(\hat{C}=\tilde{M}^\top \tilde{M}\) . If we consider each column of M to be a variable for which we have k observations, the diagonal elements of \(\hat{C}/(k-1)\) are the sample variances and. the off-diagonal elements are the sample covariances. Often the eigenvalues of \(\hat{C}\) or \(\hat{C}/(k-1)\) are called the variances.
Typically, in this set-up, the columns of the matrix are the features. Each row is considered an observation (of each of the features). \(\hat{C}/(k-1)\) is considered the empirical variance matrix of the data.
Now suppose that the SVD of \(\tilde{M}=\tilde{U}\tilde{D}\tilde{V}^\top\). \(\hat{C}=\tilde{M}^\top \tilde{M}=\tilde{V}\tilde{D}^2\tilde{V}^\top\). So the eigenvalues of \(\hat{C}\) are \(\tilde{d}_{ii}^2\) the squared singular values of \(\tilde{M}\) . And the eigenvectors of \(\hat{C}\) are the right singular vectors of \(\tilde{M}\) i.e. the eigenfeatures.
In PCA, we only compute \(\tilde{V}\) and \(\tilde{D}^2\). However, since \(\tilde{D}^2\) is diagonal, we can readily compute \(\tilde{D}^{-1}\). Thus we can readily compute \(\tilde{U}=\tilde{M}\tilde{V}\tilde{D}^{-1}\).
A commonly used plot is a plot of the data "on" two of the PCs. When we plot the data on the PCs, we are actually plotting the entries of the corresponding rows of \(\tilde{U}\). Often we do not bother to multiply by \(\tilde{D}^{-1}\) which is only a multiplicative scaling of the axes and plot the entries of the corresponding rows of \(\tilde{U}\tilde{D}\). e.g. When we plot the data on the first two PCs, we are actually plotting \((U_{i1},U_{i2})\).
There is a bit of confusion over terminology, as SVD is sometimes referred to as uncentered PCA, and PCA is sometimes referred to as the SVD of the column centered data (although PCA focuses only on the singular vectors corresponding to columns).
In bioinformatics, usually \(g >> n\) and the features are the rows of the matrix. To use PCA software such as prcomp and princomp in R, and to get the centering correct, we need to transpose the data matrix. This does not cause any computational problems (because the software actually uses the SVD to do the computation, rather than compute the empirical covariance matrix) but I find it confusing. I prefer to use SVD software and just keep track of whether I want to center the rows (for eigengenes) or columns (for eigensamples).
PCA or SVD?
PCA focuses on "explaining" the data matrix using the sample means plus the eigencomponents. When the column mean is far from the origin, the first right singular value is usually quite highly correlated with column mean - thus using PCA concentrates on the second, third and sometimes higher order singular vectors. This is a loss of information when the mean is informative for the process under study. On the other hand, when the scatterplot of the data is roughly elliptical, the PCs typically align with the major axes of the ellipse. Due to the uncorrelatedness constraint, if the mean is far from the origin, the first singular vector will be close to the mean and the others will be tilted away form the major axes of the ellipse. Thus the first singular vector will not be informative about the spread of the data, and the second and third singular values will not be in the most informative directions. Generally, PCA will be more informative, particularly as a method for plotting the data, than uncentered SVD.
"Raw" data or z-scores?
Recall that for gene expression clustering, we often prefer to use z-scores, which emphasize the up and down pattern of gene expression, rather than the magnitude of expression. Similarly, for eigen-analysis, we might prefer to use the gene-wise z-scores. When the genes are in the rows of the data matrix, this is the centered and scaled values of each row.
As an example of the use of SVD to detect patterns, we will consider the primate brain data from homework 5. These data came from a microarray study of gene expression brains from 3 humans and 3 chimpanzees, dissected into 4 brain regions before RNA extraction and hybridized to Affymetrix human microarrays. The data were normalized using RMA and we used LIMMA to compute the mean of each gene expression for each brain region. The homework stored the mean log2(expresson) for each gene for each brain region in FitTrtMeans\$coef.
Although SVD can be used with the individual samples, when there are multiple treatments and multiple replicates per treatment, we often average the expression of each gene over the replicates before doing gene clustering or finding eigengenes. For this example we used the 200 most significantly differentially expressed genes as determined using LIMMA and M=FitTrtMeans\$coef.
Recall that brain regions 1 -- 4 came from chimpanzees and 5 -- 8 came from humans. Regions 1 and 5 are Broca's region, 2 and 6 are Caudate, 3 and 7 are Cerebellum and 4 and 8 are Prefrontal cortex.
Analysis using Euclidean Distance
I used complete linkage clustering with Euclidean distance to cluster the genes, and then used cutree to create 6 clusters. The mean of the genes in each of the 4 largest clusters is shown below in the left-hand panel. (The right panel displays the clusters obtained using correlation distance and will be discussed later.) We can see that the most dominant pattern is differential expression between cerebellum and the other brain regions, although there is also a pattern of differential expression in caudate. There appear to be few differences between chimpanzees and humans in these patterns. As well, it appears that clusters 2 and 4 could be merged.
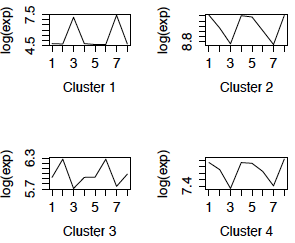
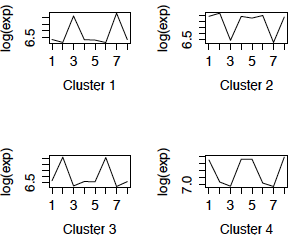
Figure 1: Cluster profiles using Euclidean distance Cluster profiles using correlation distance
We now look at patterns in the data using SVD. Our matrix M has g=200 features (genes) and n=8 treatment means (so I will call the columns of V "eigentreatments" instead of eigensamples). Notice that this implies that there are 8 eigengenes and 8 eigentreatments.
Here are the first four eigengenes plotted against brain regions. Again we see that cerebellum and caudate create most of the patterns.
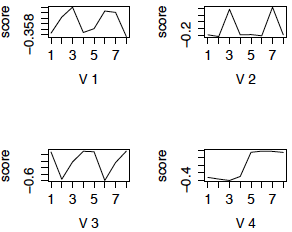
Figure 2: First 4 eigengenes using SVD
Notice that three of the the patterns are very similar to the cluster profiles, and seem to have a clear interpretation. For example, the first 3 eigengenes are genes that are differentially expressed between specific brain regions and are expressed in the same way in chimpanzees and humans. Eigengene 4 is the pattern of differential expression between humans and chimpanzees. Note however, that in SVD, unlike clustering, the direction of the pattern does not matter - i.e. in eigengene 2, all the genes that are differentially expressed in regions 3 and 7 (cerebellum) compared to the other regions contribute to the eigengene, whether they are up or down
There are several main differences between the patterns one obtains from SVD compared to finding clusters and then averaging within cluster:
1) The number of clusters of features is limited only by the total number of features. The number of clusters of samples is limited only by the total number of samples. But in SVD, there are at most min(g,n) eigenfeatures and eigensamples.
2) Clusters do not have an order of importance. The eigencomponents are ordered by the singular values. Often a the proportion of variance explained by the first few singular values is close to 1 and only the associated eigencomponents are considered important.
3) If the features and samples are clustered separated (e.g. a heatmap) there is no association between the feature clusters and the sample clusters. However, the ith eigensample and eigenfeature are associated through the decomposition.
4) Each pattern that is found in a cluster could be flipped horizontally, which would lead to another cluster. However, the eigencomponents and their mirror images are the same.
Another idea that has been used (WCGNA) is to cluster first and then use SVD within clusters. Often, however, only the first eigengene appears to be important, and it is highly correlated with the cluster mean.
Analysis using PCA with the Variance Matrix (analogous to Euclidean distance)
We now redo the SVD using the row (gene) centered data. This is also called PCA (using the variance matrix). The first 4 eigengenes are displayed in the left panel below. (The right panel are the eigengenes using the z-scores of the genes, i.e. PCA using the correlation matrix, and will be discussed later.) Notice that 3 of the 4 eigengenes are very similar to the eigengenes from the uncentered analysis (except for possible flipping to the mirror image). However, centered eigengene 3 is something new. Note that the scores for the chimpanzee brain regions are the flipped version of the scores for the human brain regions. This eigengene is an interaction of species with some of the brain regions.
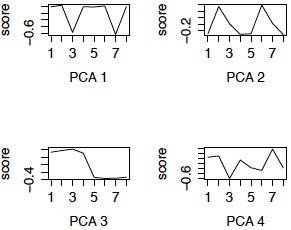
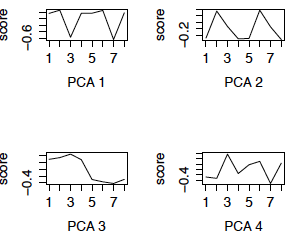
Figure 3: First 4 PCs, Centered only First 4 PCs, z-scores
Analysis using correlation distance
We now redo the analysis using the z-scores of log2(expression) for each gene. We start by looking at the cluster profiles after using hierarchical clustering with correlation distance and cutting into 6 clusters. We show the profiles of the 4 largest clusters in the right panel of FIgure 1. The missing clusters have 3 and 1 genes respectively. Again the cluster profiles appear to be based primarily on differential expression in cerebellum and caudate, with little difference between species. They are quite similar to the cluster profiles using Euclidean distance.
The eigengenes computed from the z-scores shown in the right panel of Figure 3 are quite similar to those computed from the centered data (PCA 4 is flipped) and have similar interpretations.
Comments
In this case, the eigengenes associated with the 4 largest singular values were readily interpretable. However, this need not always be the case. Often, only the first eigengene is interpretable, as the subsequence eigengenes are constrained to be uncorrelated. For example, if the two most dominant patterns in the data are correlated, then the most dominant pattern will be the first eigengene, and the second pattern will be a weighted average of the first and second eigengene.
In the case of SVD and PCA, the idea of "dominant pattern" can be made precise through the use of the singular values (SVD) or eigenvalues (squared singular values, PCA). This is the topic of the next section.
When you do the singular value decomposition the first left and right singular vectors are always the most dominant patterns in the data. However left singular vectors are all selected to be uncorrelated with one another and the right singular vectors are selected to be uncorrelated with one another. Because of this, after the first singular vector, the remaining singular vectors are not always readily interpreted as patterns in the data. For example, the next most predominant pattern might not be an eigengene - it might be a weighted average of the first 2 eigengenes. However, plotting the data on the first 2 or 3 singular values often produces an informative plot, as the clusters of features (or samples) with similar patterns will cluster together.
SVD and PCA always produce eigencomponents just as cluster analysis always produces clusters. However, unlike clusters, each eigencomponent comes with a measure of its importance - the singular values or alternatively the percent variance explained (squared singular values over total sum of squares). The cumulative percent variance explained by the first k eigencomponents is identical to \(R^2\) in multiple linear regression and is a measure of how much of the data matrix can be predicted by regressing the entries of the data matrix on k eigenfeatures or eigensamples.
Often we plot either the singular values or the percent variance explained (which is the squared singular value as a percentage of the total sum of squares) against the number of the singular vectors. This is called the scree plot. Below we show the scree plot for the SVD of the primate brain data. We can see immediately from this plot that the first singular vector is the most important. while the last few have about the same level of importance. Because we did not center the data, the first eigentreatment (which has an entry for every gene) is highly correlated with the mean expression for each gene - in fact for this example, the correlation is -0.9999. This is the dominant pattern in the eigentreatments. Comparing with the treatment-wise plots in Section 15.2, we can see that the first eigengene is approximately the contrast between the mean of expression in caudate and cerebellum compared to the other two brain regions.
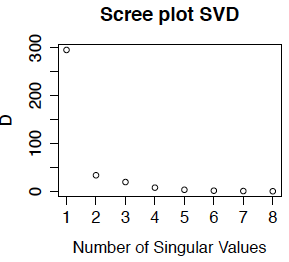
The English word "scree" refers to the pile of eroded rock that is found at the foot of a mountain, as in the picture below. We use the scree plot to pick out the mountain (i.e. the important singular vectors) from the rubble at the bottom (the unimportant singular vectors) which are interpreted as noise. Although our scree plot suggests that only one, or at most 2 of the eigengenes are important for "explaining" the gene expression data, I would probably use all 4 of these eigengenes because they all have interpretable patterns that have biological meaning.

An alternative to the scree plot is to use the cumulative sum of the percent variance explained (this could also be plotted, but that is not really necessary). In this example, the first 3 eigengenes have cumulative percent variance explained:
0.981 0.994 0.999 0.999
so 98% of the variance in the gene expression matrix is explained by the first eigengene. However, this "variance" is the average squared deviation from the origin, not from the gene means. Since the overall mean expression (over all 200 genes and 8 treatments) is about 7.2, it is not surprising that we can predict the data matrix very well with even one eigencomponent (compared to predicting all of the entries as zero).
Let \( U_1, U_2 \cdots U_n\) be the columns of U and \( V_1, V_2 \cdots V_n\) be the columns of V. It turns out that regressing the data matrix M on the first k eigenfeatures or eigensamples, is the same as computing estimating M by
\[\hat{M}=\sum_{i=1}^k U_i d_{ii} V_i^\top .\]
The regression sum of squares is \(\sum_{i=1}^k d_{ii}^2\) and the residual sum of squares is \(\sum_{i}\sum_j(M_{ij}-\hat{M}_{ij})^2=\sum_{i=k+1}^n d_{ii}^2\).
Scree Plot for Principal Components Analysis
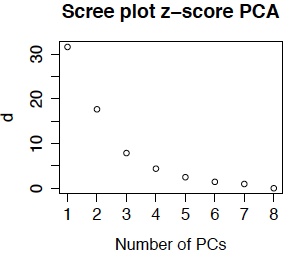
Scree plots are also used in principal components analysis. Because the feature means have been removed, the eigenvalues \(d_{ii}^2\) are now actually variances (computed as the sum of squared deviation from the gene means). Most PCA routines produce a scree plot - some plot \(d_{ii}^2\) and others plot \(d_{ii}\). The routines in R plot \(d_{ii}\) and call the singular values the standard deviations (because they are the square roots of the variances). Below is the scree plot for ordinary PCA. We see that \(d_{11}\) is much smaller than obtained from the uncentered data. This is because we are approximating the centered data matrix \(\tilde{M}\) for which the mean entry is 0 (not 7.2). We also see that the first 3 or 4 singular values are relatively large, so it is not surprising that we obtained interpretable results for the first 4 eigengenes.
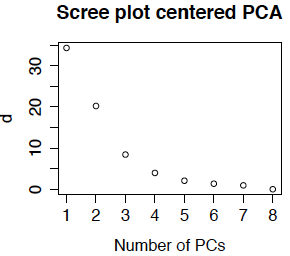
Now the cumulative percent variance explained are 70% 94% 99.4% 99.6%. The usual rule of thumb is to select enough PCs to explain over 90% of the variance, or until the scree plot flattens out. This means that we would generally pick 2 or 3 principal components, although again, because they were biologically interpretable I would likely use all 4 (or more if the 5th or 6th were also interpretable).
Another option (which is less commonly used in bioinformatics) is to compute the z-score for each gene before doing PCA. The resulting scree plot for the primate brain data is below. (It is so similar to the plot above that I double-checked to be sure that I had not recopied the plot!) The reason that the plots (and the eigentreatments) are so similar is that there is not much different in the variability from gene to gene.
SVD, PCA and Clustering
There are several ways to combine clustering with 'eigen' methods. Some methods such as WGCNA [https://labs.genetics.ucla.edu/horvath/CoexpressionNetwork/Rpackages/WGCNA/] find clusters and then use eigenfeatures within each cluster. For instance, cluster analysis is performed on all of the genes without doing any filtering on it differential expression. Everything is clustered and then an SVD is done on each cluster. Often what happens is that each cluster has one dominant eigencomponent, the cluster profile. This reassure you that the cluster has been correctly identified -if there are multiple important components it is likely that the cluster should be split.
Usually the eigenfeatures are used for plotting, i.e., feature i is plotted as \((U_{i1},U_{i2})\). This is called the plot against the first 2 principal components. We might also plot against principal components 1 and 3, and 2 and 3 if the first two principal components do not explain most of the variance. There is also a more complicated plot called the biplot which helps you determine which gene(s) have the dominant patterns. Biplots are not intuitive, but they are very informative once you have learned to interpret them.
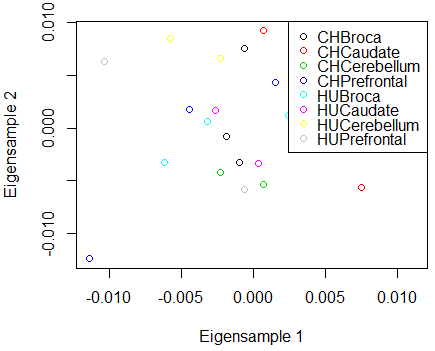
PCA is highly related to k-means clustering. Many k-means clustering packages plot the data against the PCs to visualize the clusters, as the cluster centers are often well spread out in the eigenfeature directions. The plot of the treatment means against the first two eigentreatments for the brain data is below - no interesting clusters appear to be in these data. The plot against the eigengenes is equally uninformative in this case.
Denoising using SVD
Another use for SVD is for noise reduction. Unlike differential expression analysis, SVD and PCA can find patterns in unreplicated data. After choosing the number of eigencomponents k the denoised version of M is
\[\hat{M}=\sum_{i=1}^k U_i d_{ii} V_i^\top .\]
If we start by centering the data (PCA), we also need to add back the row mean to every row. As mentioned earlier, this is equivalent to regressing the entries of M on the left or right singular values. It can be shown that for any number of predictors k the SVD reconstruction of M is the linear predictor which most reduces the residual sum of squares.
Missing value imputation using SVD
You can also use the denoised version of M to fill in any missing values by prediction. This requires an SVD algorithm such as SVDmiss in the R package SpatioTemporal which can estimate the singular values and singular vectors with missing values. We then replace any missing entries \(M_{ij}\) by \(\hat{M}_{ij}\). We tend to get better prediction if we use a small number of components k when doing the imputation, as we are then fitting the strongest patterns in the data, rather than the random components. The SVD is one of the best ways of doing imputation of missing data.
SVD, PCA and patterns in the data matrix
The maximum number of singular vectors or principal components is the same as min(g,n) - i.e. the number of samples or features. . This doesn't mean that we don't have more patterns in the data. SVD and PCA find a "basis" for the patterns - i.e. every pattern in the data is a weighted average of the singular vectors.
Other pattern finding methods using matrix decomposition
There are other 'latent structure' models that use this kind of matrix decomposition. Factor analysis is very similar to singular value decomposition and principal components analysis.
Another method, called independent component analysis ICA attempts to find statistically independent components to the data matrix. For Normally distributed data, independent is the same as uncorrelated, and this reduces to PCA. However, for non-Normal data, ICA may find other interesting structure.
Machine learning has given us another matrix decomposition method called nonnegative matrix decomposition that decomposes M as a produce of the form
\[\hat{M}=AB^\top\]
where A is an n × k matrix and B is a k × k matrix both of which have only nonnegative entries (i.e. every number in the matrices is zero or positive). Compared to using SVD to predict or denoise M, the resulting \(\hat{M}\) is often quite similar. However, the entries of A (the eigensamples) and B (the eigengenes) are all nonnegative, which is compelling when the original data matrix M has only nonnegative entries since we would not want to have negative expression. The resulting matrices may not be unique but often have properties had enhance interpretability, such as having many zero entries.
Finally, ordination methods can be used to find axes which spread out the data as much as possible. For example, we could use the Euclidean distance as a measure of distance between samples (using our g genes) and then try to find a much smaller set of variables which give us almost the same distance matrix - these variables are called ordination axes. They will have a co-ordinate for every sample, and so they have the dimension of eigenfeatures. Multidimensional scaling is a popular ordination method which is sometimes referred to a nonparametric PCA.
SVD and PCA are called unsupervised dimension reduction because the act only on the data matrix. Often as well as our feature by sample matrix, we have other information about the samples such as phenotypes, population subgroups and so on which we want to predict from the feature by sample matrix. Since we have many more features than samples, in order to do this in a meaningful way we need to reduce the number of features (i.e. dimension reduction). One way to do this is to use the first few eigenfeatures as predictors. This is called principal component regression. However, because determining the eigenfeatures uses only the sample matrix and not the other information, there is no guarantee that these eigenfeatures will be good predictors.
Methods that use the response variable to develop predictors are called Sufficient Dimension Reduction or SDR methods. When the response variable is categorical (such as brain region) the simplest dimension reduction method simply averages the samples over each category as we did to find the eigenfeatures and eigentreatments. When the response variable is continuous such as milk production or biomass we create categories by "slicing" the response variables into bins - e.g. weight groups. Besides simply averaging the data within bin or category and doing SVD, there are a number of more sophisticated methods such as Sliced Inverse Regression (SIR) and Sliced Average Variance Estimation (SAVE), which find features useful for prediction. One interesting finding with such methods is that they can find features that are useful for nonlinear as well as linear regression.
Full discussion of SDR methods is beyond the scope of this course. However, those interested can find information in https://projecteuclid.org/euclid.ss/1185975631 [1].
Links:
[1] https://projecteuclid.org/euclid.ss/1185975631