1.5 - Gene Expression
The first half of this course will focus on gene expression and its measurement using microarrays and sequencing.
As mentioned earlier, the chromosomes are storage devices made out of DNA (except a few organisms which use RNA). The active molecule is RNA, which is based on the DNA as a template. Some of the RNAs have their own function. Some RNAs called messenger RNAs (mRNA) are used as templates for protein production. mRNAs are called coding RNAs. The functional non-coding RNAs are called ncRNA.
Transcription is the process by which the DNA template is used to create a complementary RNA. Many RNAs then go through post-processing steps such as folding for ncRNAs or splicing (removal of introns) for mRNAs.
Transcription begins with proteins called transcription factors binding to the promoter region upstream of the gene. There are complex chemical processes involving things like the coiling structure of the DNA and methylation that enhance or dampen the process. Once transcription starts, the DNA unzips and RNA is matched using one strand of the DNA as a template. After post-transcriptional processing, relatively stable transcripts are created. Because of the simple chemical structure of DNA and RNA, and with the use of an important biochemical process called reverse transcription, it is relatively simple to determine the sequence of the DNA and RNA and to quantify the transcripts. Microarrays and high-throughput (massively parallel, next generation) sequencing are both methods which capture, identify and quantify pieces of DNA.
Here is a very simplified picture of the transcription process.
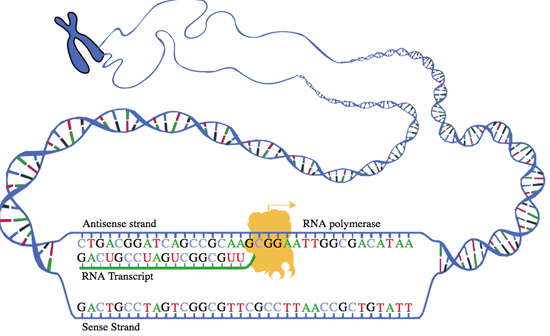
Transcription starts when a transcription factor finds the promoter region and this initiates the process of transcribing each base in the DNA to a matching RNA. This is called the pre-RNA because it is not in its final form. This transcription process always begins at the start codon at 5' of the gene and goes towards the 3' end until a stop codon is reached. (A codon is a set of three bases. For coding RNAs, each codon is either a start, stop or matches a single amino acid, a basic building block of a protein.)

For coding RNA, the pre-RNA contains both the introns and the exons. In post-transcriptional modification, the introns and possibly some exons are excised and the mRNA is created. These exons can be put into many different combinations to form different isoforms (also called splice variants). For most coding RNAs, a string of "A" nucleotides is attached to the 3' end and is called the"poly-A tail".
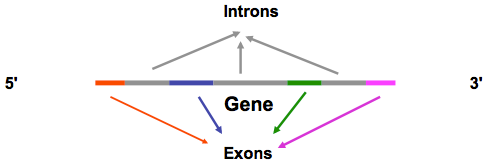
A gene that has introns can make several different transcripts based on which exons are used. Different splice variants of a single gene may encode different proteins or have different regulatory regions that determine how they are used in the cell. For example, in humans different isoforms of the haemoglobin protein are expressed during fetal development and as an adult. Expression of the wrong isoform at a particular developmental stage or in a particular tissue can lead to disease.

The poly-A tail is useful to biologists because it can be used to chemically retrieve transcripts by matching the tail. Since a lot of RNA fragments not of interest may be in a sample, the ability to retrieve only the interesting transcripts is very important. On the other hand, use of the poly-A tail to retrieve transcripts means that transcripts without poly-A tails, including some mRNAs, may be systematically missing from the samples.
ncRNAs also come from post-transcriptional processing which may including dicing the pre-RNA to obtain the shorter functional fragments, and possibly folding or other structural changes. ncRNAs do not have a poly A-tail.
For mRNA, the next biological step involves the transcript being turned into a protein. This is called translation. In many cases we would prefer to measure the proteins in the cell, but they are much more chemically complex and hence harder to quantify. Once they are quantified, however, the statistical methods used for their analysis is very similar to methods for analysis of nucleic acid data.
Questions that researchers ask about RNA include:
- What genes are expressing RNA in a tissue?
- What splice variants are being expressed in a tissue?
- What differences are there in RNA expression between conditions (e.g. different tissues or under different treatments)?
- What biological process (e.g. methylation, protein or ncRNA binding) is turning the genes on and off?
Many of these items will be covered in more depth in this course, especially the statistical analyses for these procedures.

