1.10 - Massively Parallel Sequencing
Around 2005, new methods for very fast, accurate DNA sequencing became available. This means that we can also sequence cDNAs. These next-generation technologies can read the genomic sequences of millions of fragments of DNA. Interestingly enough, you don't need to know anything about the organisms, and a priori sequence information is not required. The most recent technologies allow sequencing of RNA directly, without conversion to cDNA. Although these methods are currently not as high throughput as DNA sequencing, technological advances are very rapid in this area, increasing throughput, accuracy and the lengths of the fragments that can be sequenced.
The cost of sequencing has come down considerably, although sample preparation is still somewhat expensive compared to microarray sample prep. However, the biggest problem is transporting and storing the raw sequence data. Sequencing data sets run anywhere from 10 to 15 Gb. Some of these data sets are so large that it is faster and cheaper to load up an external hard drive with terabytes of data and ship it than it would be to rely on downloading this over the Internet. Long-term storage of the data is also problematic.
The newest technologies will give you 1 - 250 million fragments (reads) per sample. Shorter fragments are cheaper per read, but are also less informative. Most of the technologies allow a variety of read lengths from about 50 to a few hundred nucleotides. In general, technologies that allow longer reads are more expensive per nucleotide than those that have shorter maximum read length.
Here is some data from Marioni et al., 2008. This is the raw data. The reads were oligos of length 36 (36-mers). The output is stored in a FASTA format or FASTQ (which also has quality scores).
GGAAAGAAGACCCTGTTGGGATTGACTATAGGCTGG
GGAATTTAAATTTTAAGAGGACACAACAATTTAGCC
GGGCATAATAAGGAGATGAGATGATATCATTTAAG
When these data were collected, read lengths of 50 bases were considered long. Now, the reads are longer. 100 bases is usually adequate for gene expression in sequenced organisms which have a good reference genome or transcriptome. Much longer reads are desirable for assembling a genome or transcriptome or for complex genomes which have duplications such as tetraploids.
The strings of characters need to be identified. When there is a reference genome or transcriptome, this is usually done by mapping - i.e. matching the sequences to sequences (or complementary sequences). When there is no reference, the reads are matched to one another to build a de novo assembly which is then used as the reference.
Sequencing is very good at the following tasks with DNA:
- "de novo" sequencing
- metagenomics i.e. sequencing mixed communities of multiple species
- resequencing (detecting biological variation in a population)
- SNP detection and quantification (detecting biological variation in a population)
- protein binding site discovery and quantifying binding
- methylation site discovery and quantifying methylation
- chromatin interaction (detecting regions of chromosomes that interact)
- and anything that you can think of that measures a piece of DNA, including creating micro arrays.
Sequencing is also very good at the following tasks with RNA:
- quantifying gene expression
- quantifying exon expression
- quantifying non-coding RNA expression
- isoform discovery
- quantifying isoform expression
- microarray probe construction
Fragmenting the (c)DNA can introduce biases. If you use a certain enzyme it might preferentially chop up the molecules at certain sites. Therefore, since the sequencer starts at one end, all the reads from that fragment contain that same bias. If you use physical force (e.g sonication) to fragment the molecules then there may be weak sites and they would preferentially break. You might think that you have higher expression or higher methylation at these loci when in fact you'll see more of these because this is where the molecules break. This is not the main focus of what this class intends to cover, but you need to be aware of this problem. The newer technologies that do not fragment the RNA or DNA have fewer biases.
Right now there are two choices for short sequencing technology: single and paired end. In single end sequencing the sequence of the fragments is determined by a sequencer starting from either the 3' or 5' end of the fragment. Paired end technology sequences from both ends possibly leaving an unsequenced link in the center. When read lengths were short, paired end sequencing was a good option to obtain longer, more informative fragments. Now that read lengths can be quite long, there may be no unsequenced link - in fact, the center of the read might be sequenced from both ends. For this reason, paired end sequencing is becoming less popular. As they become less expensive, whole molecule sequences will likely supercede short sequence technologies.
Reads are usually not useful unless we can identify them in some way. Both for mapping and assembly, longer reads are easier to use. Firstly, with longer reads the matching software can be instructed to tolerate more mismatches. Mismatches occur due to heterozygosity, differences between the reference and the samples, PCR errors and sequencing errors. Secondly, most genomes include repeat elements and gene duplications which create highly similar sequences - longer reads are less likely to match multiple locations on the reference or create a false assembly due to joining fragments from different genomic regions.
Statistically, for most of the analysis we do we will won't care about any of this. We will be talking about the data AFTER mapping. However, when you are planning a study, you need to carefully determine the appropriate sequencing length and single/paired end technology to ensure sufficient high quality data for your purposes.
The methods of sequencing differ among the 4 or 5 different types of technologies being used but they mostly have a similar flavor. The fragments are captured. PCR is used form a cluster from each fragment, and the sequencer starts reading from the end of the cluster. A chemical process is used to expose and tag the nucleotide at the end of the fragment and then the entire plate is captured as a photo at the tag frequeny. (The raw data are therefore 4 photos at each location on the fragment.) The exposed base is then stripped off and a new base is exposed.
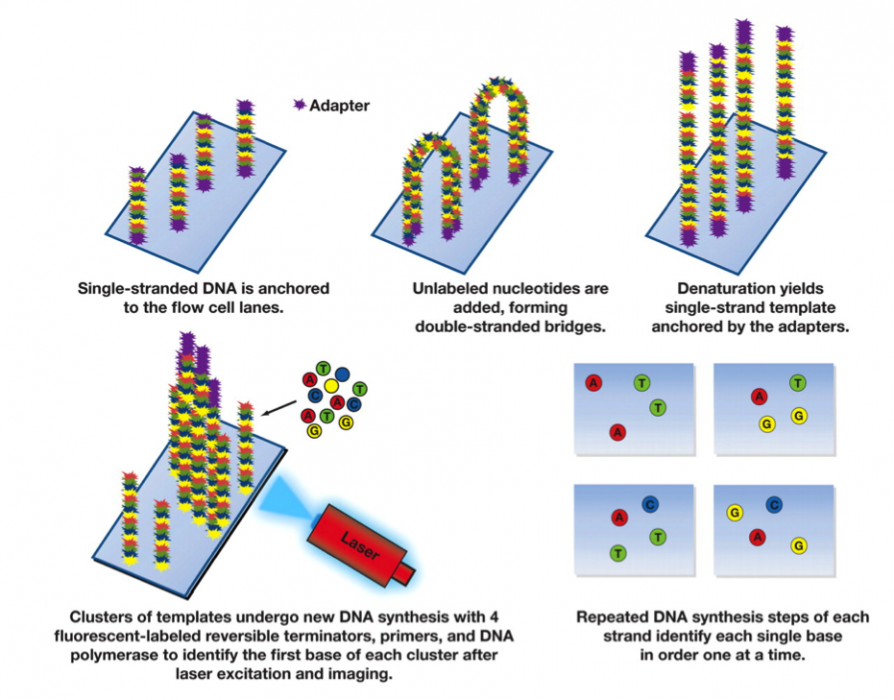
(Image used with permission from the America Journal of Clinical Pathology)
If the read length is 100, there will be 100 × 4 photos from this sample. This is because at each step a fluorescent label is added that is specific to one of G, C, A or T. Each photo therefore has a bright spot at the currently labeled clusters. In a high quality "base call" only one of the 4 nucleotides is detected in a cluster in a particular read position. In the graphic below you will see peaks of fluorescence for each of the different bases for several clusters. The peaks shown below are all high quality, because only one nucleotide is fluorescing at each location. But recall that each cluster is made up of thousands of identical strands. As the process proceeds, errors may occur because the reagents are degrading or because the already sequenced nucleotides are not detaching from the strand. As a result, the clusters may gain signal for several different nucleotides, which would show up as peaks for several nucleotides, instead of one. The quality score summarizes this. Usually the quality score decreases for nucleotide locations further down the strand. When the score is too low we often trim the reads by truncating to a smaller number of nucleotides per read.
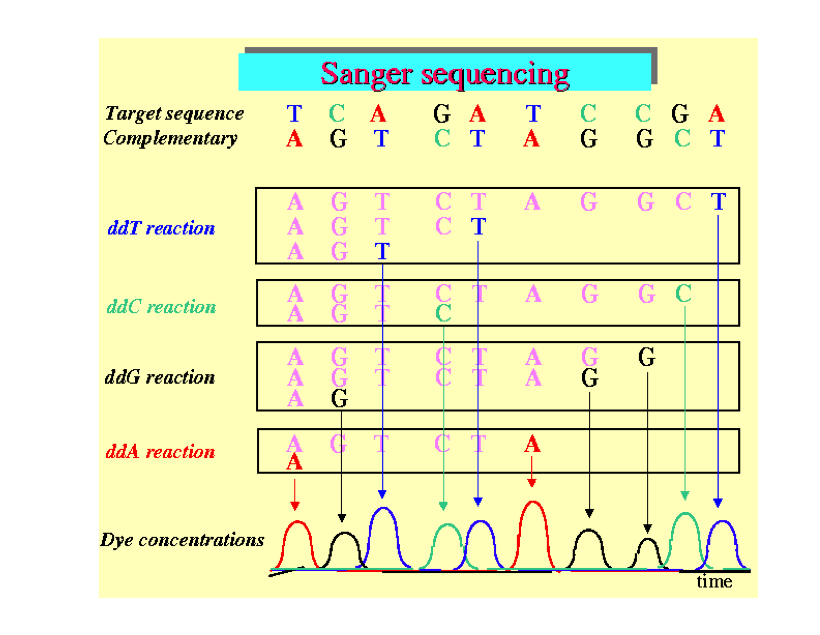
re-size image (copyright needed?)
This vector of quality scores for each base would add considerably to the size of the file. Usually all that recorded is the most likely nucleotide and the quality score for that nucleotide. Recall that the typical size of the raw data file for single sample is about 10Gb. If the data for all 4 nucleotides is retained, that increases to 40 Gb. It is often less expensive to resequence the sample than to store all the data.

